I took that course at the beginning of 2024. notes are not organized. A course from Medical Device HQ Course Link: https://medicaldevicehq.com/introduction-to-working-in-the-medical-device-industry-online-course/
In USA, we have 21 CFR 820, in EU we have (MDR) Medical Device Regulation of EU
PMA 510(k) and Device listing. PMA is most expensive and time consuming option
Audit performed by external client should be referred as second party audit
CE-marked medical device in EU needs a QMS and Technical documentation
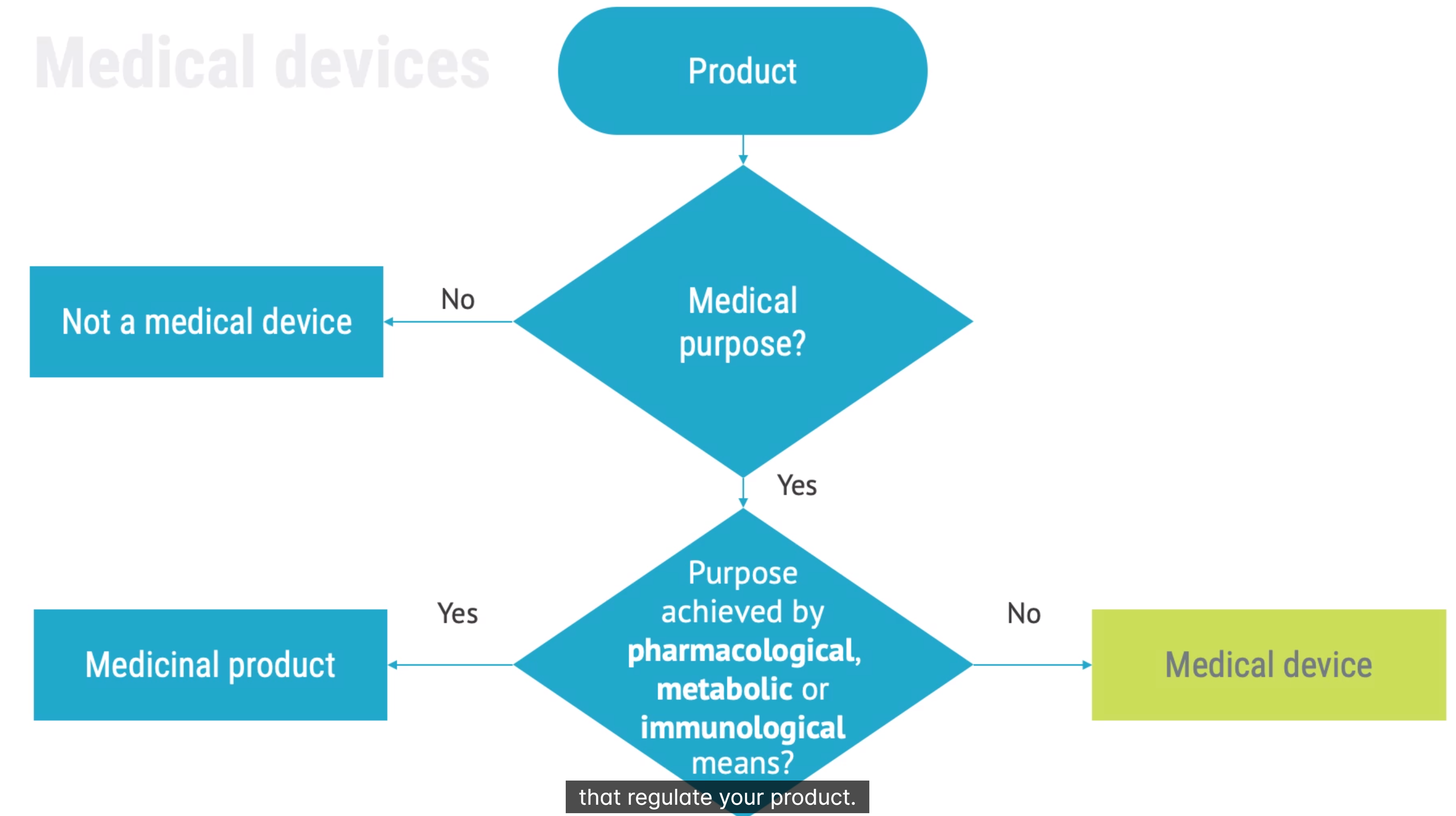 diagnosis therapy surgery or birth control. achieves primary purpose not by pharmacological metabolic or immunological
diagnosis therapy surgery or birth control. achieves primary purpose not by pharmacological metabolic or immunological
combination products: medical product with medicinal product The legal manufacture is responsible for describing intended use of a product. Intended Use of Medical Device
Medical Device Terminology
(QMS) Quality Management System: QMS, All manufacturers should have QSR and ISO 13485, can be paper or digital
Quality of Medical Device: about fullfilling requirements, not only product but also system must be quality
Legal Manufacturer of Medical Device: responsible of design and manufacturer. They can outsource real manufacturing but whoever has design and qms is legal manufacturer
Authorized Representative of Medical Device: a person in EU for MDR, US FDA agent for USA scenario
 (MDR) Medical Device Regulation of EU is focused but we also have IVDR
(MDR) Medical Device Regulation of EU is focused but we also have IVDR

Standards
Without standards, you should reinvent wheel.
 there is product standarts and process standarts Product Standards vs Process Standards
ISO 13485 is QMS. Applied to medical device manufacturers depending on classification
ISO 14971 for risk management for medical devices. It shall cover all the lifetime of product.
IEC 62366-1 human factors engineering close relationship with ISO 14971
ISO 10933-1 biocompatibility. It applies to all contact with people. It is not applicaple to SW.
IEC 60601-1 safety and essential performance for medical electrical equipment
IEC 62304 for software life cycle process
there is product standarts and process standarts Product Standards vs Process Standards
ISO 13485 is QMS. Applied to medical device manufacturers depending on classification
ISO 14971 for risk management for medical devices. It shall cover all the lifetime of product.
IEC 62366-1 human factors engineering close relationship with ISO 14971
ISO 10933-1 biocompatibility. It applies to all contact with people. It is not applicaple to SW.
IEC 60601-1 safety and essential performance for medical electrical equipment
IEC 62304 for software life cycle process
Introduction to Audits
audit will find you 🙂
if audit goes wrong, you can lose the right to manufacture
competent authorities = fda
Medical products agency in SE
Every country have compenent
Notified Bodies of MDR do visiting while FDA do it by themselves


Desktop audit → “Procedure vs Standard” On-site audit → “Practice vs procedures”
It can take up to 10 days
- 1st party audits = means do it yourself, internal audits
- 2nd party audits = external provider audit,
- 3rd party audit = accredited audit

Quality concepts and audits
Question areas from auditors:
- Quality in general
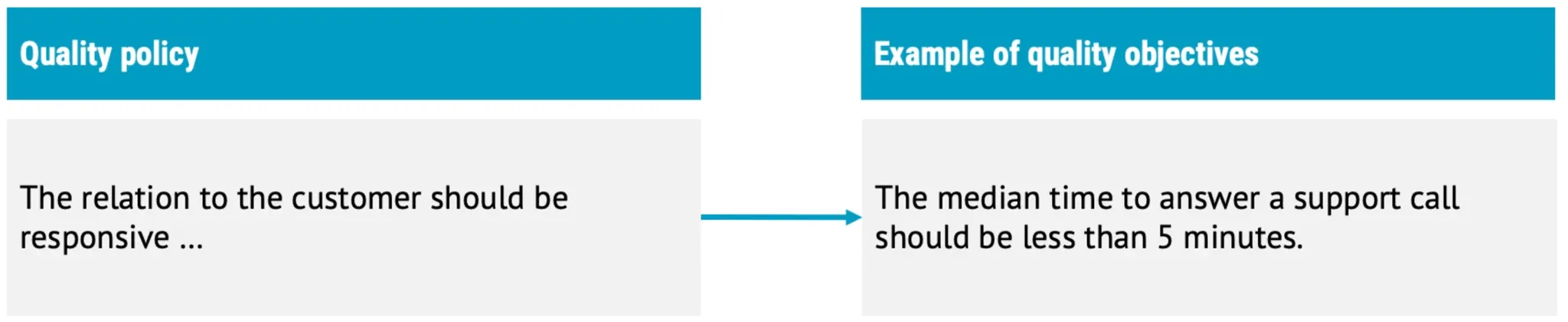
- Quality policy
- Quality objectives
- your position / role
- Responsibilities and authorities
- Observing work
- How you carry out your work
- What are the things you are using
- Records
Quality Policy of Medical Device

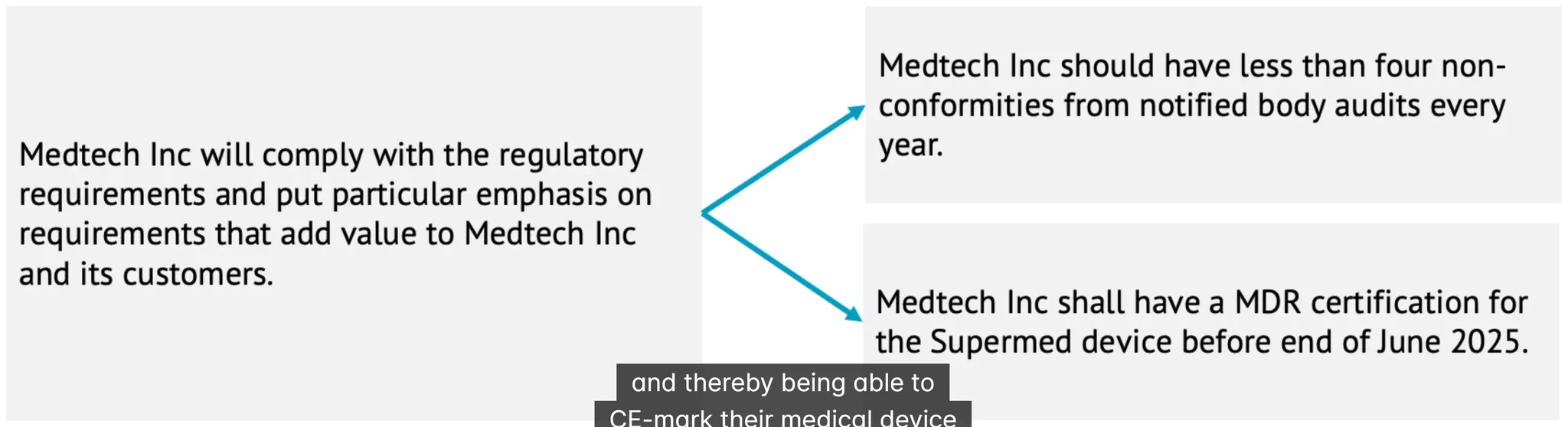
what is non-conformity

unannounced audits
it is sth in eu every 3 to 5 years, they can come. notified bodies if they come, verify that they are notified body they should carry id card to prove that
Complaint of Medical Device:
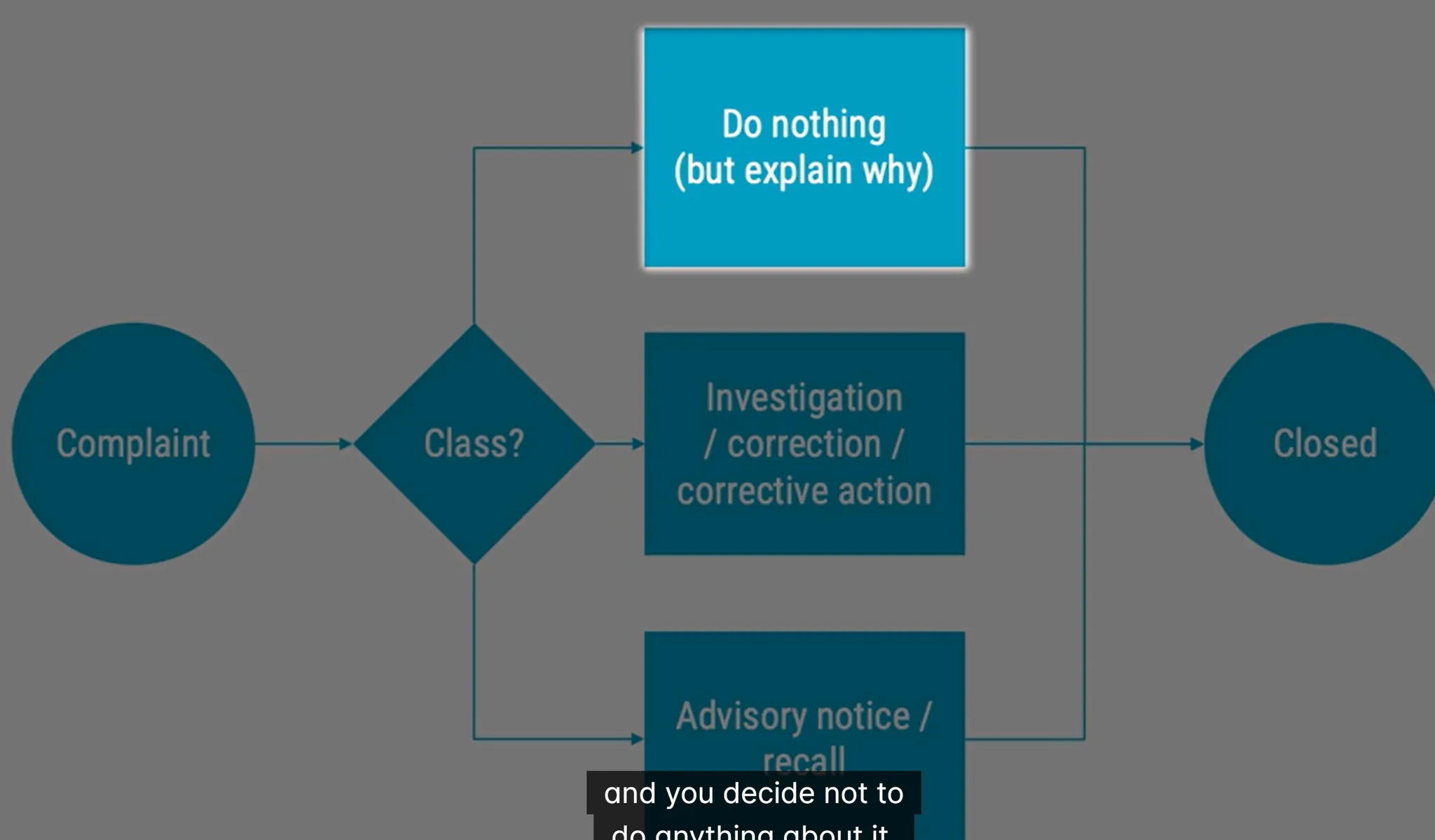 if they ask something irrelevant like why it is too heavy. then you can explain why but do nothing as it has nothing to do for regulations
written, electronics or oral that alleges deficiencies about identity, quality, durability, reliability, usability, safety or performance
they can say I am happy but it can be better
regulatory time limits if someone injured.
if they ask something irrelevant like why it is too heavy. then you can explain why but do nothing as it has nothing to do for regulations
written, electronics or oral that alleges deficiencies about identity, quality, durability, reliability, usability, safety or performance
they can say I am happy but it can be better
regulatory time limits if someone injured.
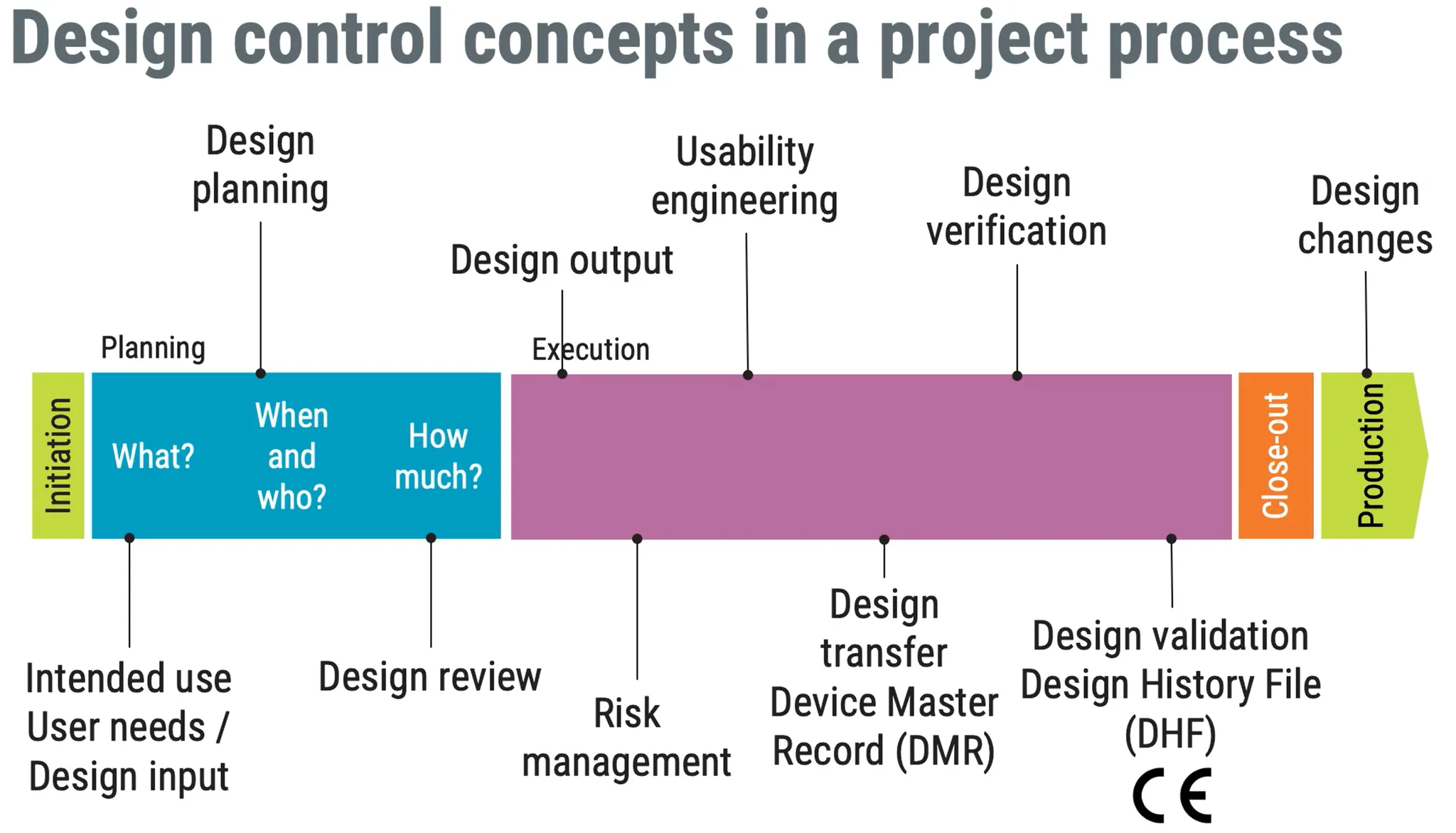
Product development
pharma industry focus on manufacturing while medical focus on design planning first= what when and who and how much? execution you should include intended use to planning
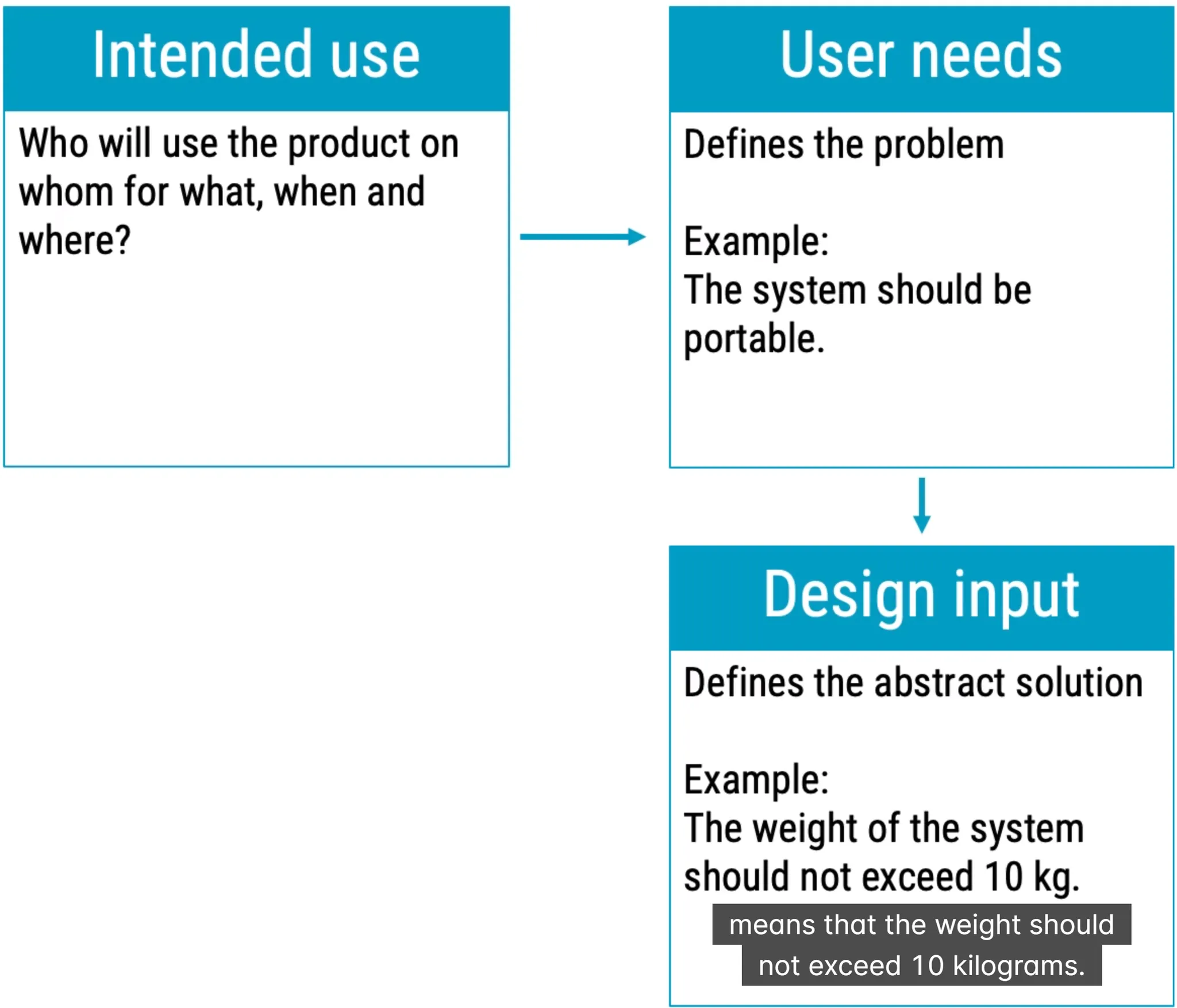 Design Review of Medical Device focused on quality and quality only
Design Output of Medical Device
risk management of product. not project
design transfer
(DMR) Device Master Record of Medical Device
then Design Verification
Design Review of Medical Device focused on quality and quality only
Design Output of Medical Device
risk management of product. not project
design transfer
(DMR) Device Master Record of Medical Device
then Design Verification
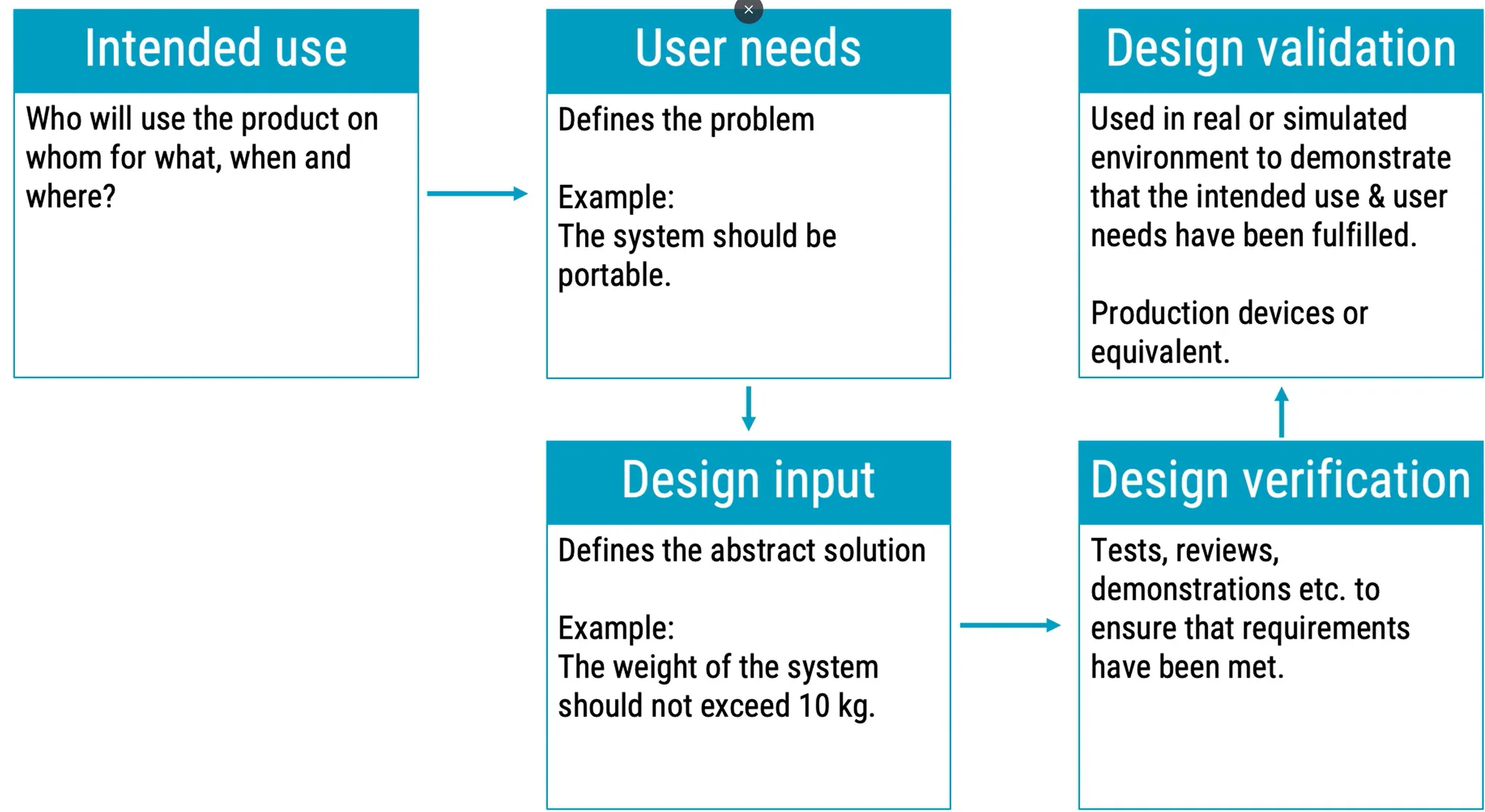
Validation of Medical Device means to check if it is really working and useful
(GSPR) General Safety Performance Requirements
you have a checklist like that:
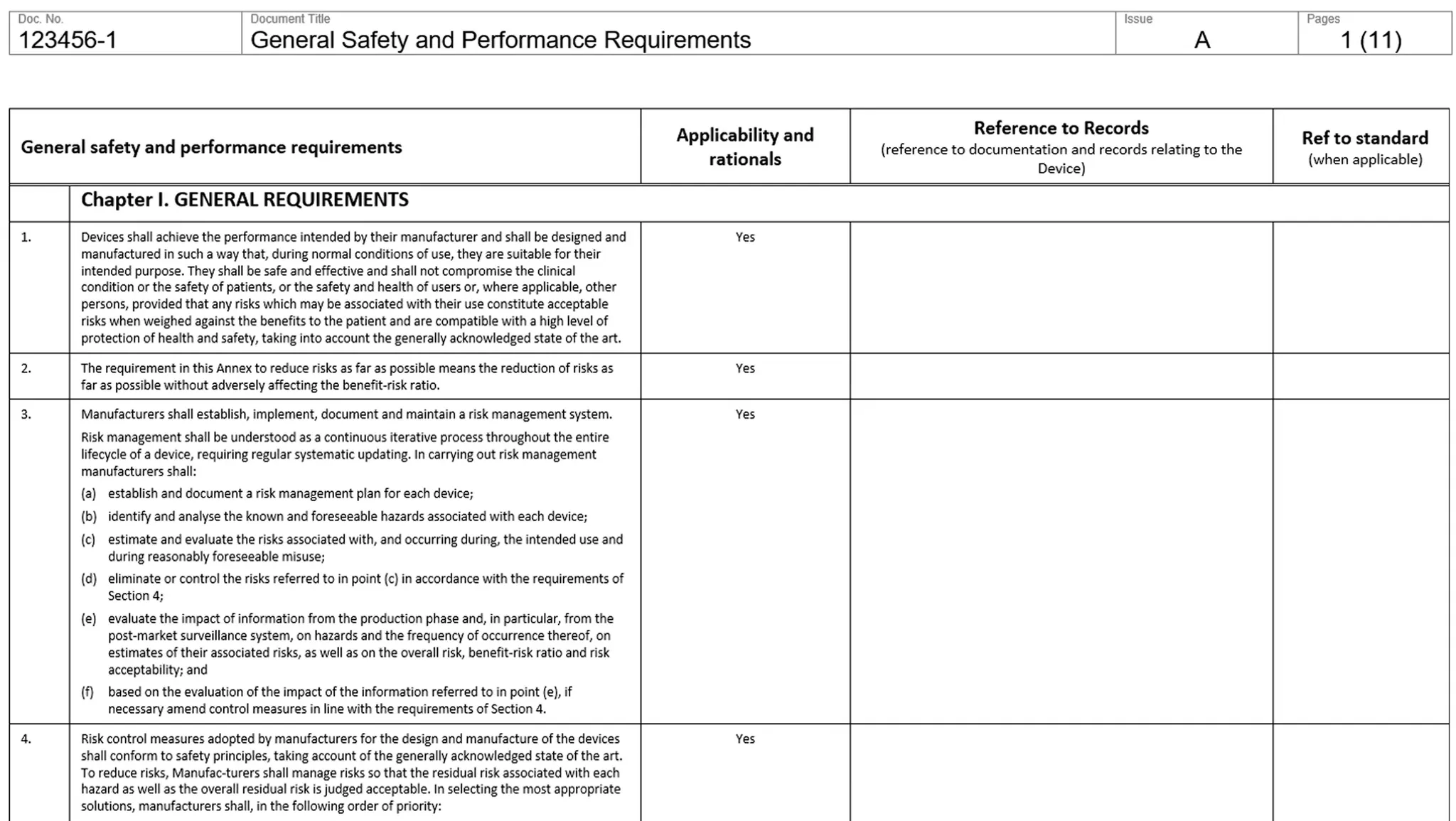
Product Documentation
(DHF) Design History File of Medical Device: rather to show what is done historically important collection of documents to auditor (DMR) Device Master Record of Medical Device How to produce the device. Shall be readily accessible. An index is usually needed due to the amount of document (DHR) Device History Record of Medical Device Contains or references the records necessary to demonstrate that device is manufactured in accordance with the approved device master record and the req of this part
Risk Management of Medical Device
for product safety, not project or company mentioned in 14 places in ISO 13485 Areas that require risk management:
- Software and proces validation
- Verification off effectiveness of training
- Product development
- Production
- Change control
- Purchasing
- Verification of purchased products
- Post-market surveillance
- Clinical investigations
Safety of Medical Device is freedom from unacceptable risk
Hazar Traceability Matrix of Medical Device


Risk of Medical Device according to ISO 14971: Combination of the probability of occurrence of harm and the severity of that harm. money or being late in project is business risk, not medical device risk :) Risk Control options:
- Inherent safety by design and manufacture (most effective)
- Protective measures in the medical device itself or in the manufacturing process (automated tests)
- Information for safety, and training to users (user experience) Safety of Medical Device means what is generally accepted. State of the art.
Usability Engineering
IEC 62366
it also means human factor engineering at fda
regulatory point of view, it only relates to safety. user friendly is not a topic of regulation
good usability sometimes need less user-friendly
user interface is not only screen, but also label papers and everything user can interact

Formative Evaluation of Medical Device
formative evaluation means same thing as in teaching this quizzes are also formative evaluation any survey to understand how user use your device
Summative Evaluation of Medical Device
summative evaluation is not the same as design validation design validation is for user needs are met, while summative evaluation checks user interface is safe
Production and Infrastructure
keep things clean and sterile production and prototype should be in different rooms, prototype should be clearly marked as prototype how to get into production and how to behave there esd can also injure components a lot of devices are invasive sterilization is made by radiation or etylene oxide sterile means packaged after sterilization tyvek pouch is perfect material for packaging dont touch your face avoid farting 🙂
keeping sterile and nonsterile product in seperateplace is regulated by ISO 13485
Product Approval
under (MDR) Medical Device Regulation of EU
you need to CE mark for sure
QMS is surely needed ISO 13485
annex 1 of mdr as GSPR
technical documentation, annex 2 of mdr
notified body
clinical evaluation and investigation
Clinical Evaluation of Medical Device vs Clinical Investigation of Medical Device

under FDA
21 CFR 820 for QMS (DHR) Device History Record of Medical Device, (DMR) Device Master Record of Medical Device Clinical Trial of Medical Device and FDA submission and inspection Device listing is a route to us market 510(k) / pre market notification Pre market approval 510(k) gives not approval but clearance
post market surveillance
monitor to safety
requirement for all in EU and most of the device in US
 Post market clinical follow-up PMCF focuse more on clinical situation
periodic safety update reports PSUR
Post market clinical follow-up PMCF focuse more on clinical situation
periodic safety update reports PSUR